DNA Analysis with Python
for Biologists
Build an Open Reading Frame finder to translate DNA sequences into proteins
🧬Today's Focus
String Manipulation
Learn to work with text data, perfect for DNA and protein sequences
DNA Sequence Analysis
Apply Python skills to real biological problems like GC content calculation
Pattern Recognition
Find motifs, restriction sites, and other important sequence features
🐍Core Python Skills
Python dictionaries
Store and manipulate biological data efficiently
String Methods
Essential tools for sequence processing and analysis
Conditional Logic
Make decisions in your code based on biological criteria
🔧Additional Topics
Python Development Tools
IDE setup, debugging, and best practices for scientific computing
Open Source Software
Understanding the ecosystem of biological analysis tools
Biopython
Introduction to the most popular Python library for bioinformatics
Today's Goal: Build an ORF Finder 🧬
Our Python Program Will:
Find Start Codons
Locate all ATG positions
Find Stop Codons
Scan for TAA, TAG, TGA in-frame
Extract ORFs
Get sequences between start & stop
Find Longest ORF
Identify the most likely protein
Translate to Protein
Convert DNA codons to amino acids
Process Many Files
Automate for 100s of sequences
Skills we'll learn: String operations • Dictionaries • Conditionals • File I/O • Biopython
Meet Darren: A Biologist with a Problem 🧬

Darren, PhD student studying gene expression
The Challenge
Darren has sequenced hundreds of mRNA molecules from cancer cells. Each sequence could encode important proteins, but finding them manually takes hours per sequence!
📁 Current situation:
- • 500+ mRNA sequence files
- • Each needs to be checked for ORFs
- • Manual checking takes ~30 min/file
- • That's 250 hours of tedious work!
💡 Solution: Automate with Python!
What takes 30 minutes by hand can be done in milliseconds with code
What are Open Reading Frames (ORFs)?
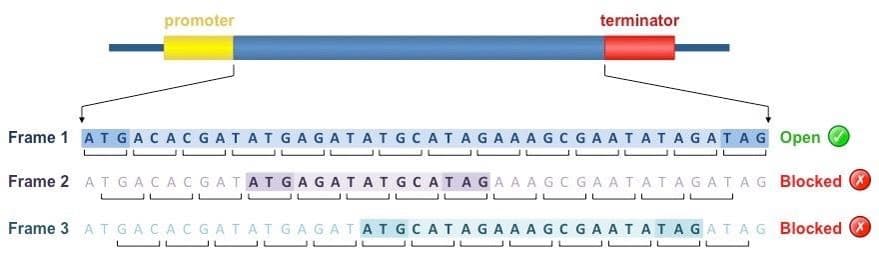
Key Concepts
- •DNA/RNA can be read in 3 different frames
- •Each frame groups nucleotides into different codons
- •An Open frame has no early stop codons
- •A Blocked frame hits a stop codon quickly
ORF Requirements
Start:
ATG (codes for Methionine)
Stop:
TAA, TAG, or TGA
Valid ORF:
ATG → ... → Stop (in same frame!)
In the example above: Only Frame 1 is "open" - it can produce a full protein. Frames 2 & 3 hit stop codons immediately!
Breaking Down the ORF Problem
To find and translate an Open Reading Frame, we need to solve 3 simple steps:
Find First ATG
Scan the DNA string and find the first ATG start codon
↑ position 6
Extract ORF
From ATG, collect codons until we hit a STOP codon
Translate to Protein
Convert each codon to its amino acid using the genetic code
💡 Our Learning Path
The Complete Code - Live Demo!
Here's our complete ORF finder - try it with the example DNA sequence!
Part 1
Python String Fundamentals
for Biology
Working with DNA sequences as strings
🎯 Our First Function: Finding the Start Codon
This is what we'll build together in the next few slides:
def find_atg(dna_sequence): """Find the first ATG start codon in the sequence.""" for i in range(len(dna_sequence) - 2): if dna_sequence[i:i+3] == 'ATG': return i return None # Return None if no ATG foundDon't worry if this looks complex - we'll build it step by step!
Quick Review: Data Types & Strings
Basic Data Types
int→ 42, -17, 1000Whole numbers
float→ 3.14, -0.5, 2.7e-8Decimal numbers
str→ "ATCG", 'DNA'Text sequences
bool→ True, FalseLogical values
String Operations We Learned
dna = "ATCGATCG"
len(dna)→ 8dna[0]→ "A"dna[0:3]→ "ATC"dna + "TAA"→ "ATCGATCGTAA"dna * 2→ "ATCGATCGATCGATCG""AT" in dna→ TrueRemember: In Python, strings are sequences of characters - perfect for representing DNA, RNA, and protein sequences!
DNA String Slicing
🔪 Understanding String Slicing
String slicing lets you extract parts of a string using [start:end] notation:
- •
string[1:4]gets characters at positions 1, 2, and 3 - • Position counting starts from 0
- • The end position is not included
🎯 Try It Yourself!
Complete the challenge: Print the last three bases of the DNA sequence using slicing.
🎯 Practice Challenge
Try these basic slicing exercises:
- • Extract just the middle 4 bases of "ATGCGTAAA"
- • Get the first half of the DNA sequence
- • Extract every other base using step slicing [::2]
- • Practice using negative indices to get sections from the end
🚀 Practice More in Google Colab!
Open the full string manipulation notebook with exercises and solutions
Finding ATGs: Loops + Slicing
🔄 Step 1: Loop Through Every Position
To find ATGs, we need to check every possible position in the DNA string:
- • Position 0: Check bases 0-1-2
- • Position 1: Check bases 1-2-3
- • Position 2: Check bases 2-3-4
- • And so on...
🔪 Step 2: Extract 3 Bases from Each Position
At each position, slice out exactly 3 bases to check if it could be a start codon:
🚧 We've Hit a Problem!
What we can do: Extract 3-base sequences from every position ✅
What we can't do yet: Check IF a sequence equals "ATG" ❌
We need: A way to make decisions in our code - Python conditionals!
📚 Coming Up Next: Python Conditionals
To solve our ATG-finding problem, we'll learn:
- •
ifstatements for making decisions - • Comparing strings with
== - •
elifandelsefor multiple conditions - • Putting it all together to find ATGs automatically!
🚀 Practice String Manipulation in Google Colab!
Try the full string manipulation notebook with more loop and slicing exercises
Part 2
Python Conditionals
Making Decisions
Teaching Python to make choices based on biological data
🔍 Focus: The Conditional Logic
Notice the if statement that makes the decision:
def find_atg(dna_sequence): """Find the first ATG start codon in the sequence.""" for i in range(len(dna_sequence) - 2): if dna_sequence[i:i+3] == 'ATG': # ← The key decision! return i return None # Return None if no ATG foundWe'll learn how if statements help us find biological patterns!
Conditionals: Making Decisions in Code
🎯 Basic if Statement
Use if to make decisions based on conditions:
🔄 if-else: Choose Between Two Options
Use else to handle the opposite case:
🎪 elif: Multiple Choices
Use elif to test multiple conditions:
🔑 Key Points to Remember
🚀 Practice Conditionals in Google Colab!
Open the comprehensive conditionals notebook with exercises and biological examples
Building Our First Function: find_atg()
🎯 Goal: Find the First ATG in a DNA Sequence
Our function needs to:
- • Look at every position in the DNA string
- • Check if the 3 bases starting at that position are "ATG"
- • Return the position when ATG is found
- • Return None if no ATG exists
📍 Step 1: Loop Through Each Position
We use range(len(dna_sequence) - 2) to avoid going past the end:
🔍 Step 2: Check if Three Bases Equal "ATG"
At each position, we extract 3 bases and compare with "ATG":
↩️ Step 3: Return the Position When Found
As soon as we find ATG, we return its position and stop searching:
🧩 Function Components Breakdown
def find_atg(dna_sequence):Define function with one parameter
for i in range(len(dna_sequence) - 2):Loop through valid positions
if dna_sequence[i:i+3] == 'ATG':Check if 3 bases equal ATG
return iReturn position and exit
💡 Try It Yourself!
Modify the function to find ALL ATG positions (not just the first):
🔑 Key Concepts We Combined
Part 3
Python Dictionaries
Match Codons with Amino Acids
Using key-value pairs to store and look up biological information
🗂️ Next Function: The Codon Reader
Notice the dictionary lookup that finds stop codons:
# Step 2: Extract ORF from ATG to STOP codondef find_orf(dna_sequence, atg_index): """Find the ORF starting from ATG position until stop codon.""" orf = '' for i in range(atg_index, len(dna_sequence) - 2): codon = dna_sequence[i:i+3] if len(codon) == 3: # Make sure we have a complete codon orf += codon if codon in CODON_TABLE and CODON_TABLE[codon] == '*': # ← Dictionary magic! break return orfWe'll learn how CODON_TABLE[codon] looks up amino acids instantly!
Dictionaries: Key-Value Pairs for Biology
🗂️ Creating a Simple Codon Dictionary
Dictionaries map keys to values. Perfect for codon → amino acid!
🔍 Accessing Keys and Values
Get values by key and loop through the dictionary:
✏️ Adding and Changing Values
Modify existing entries or add new ones:
🛡️ Safe Operations: get() and pop()
Handle missing keys safely and remove entries:
The Two Core Functions - Interactive Demo
📚 Dictionary & Functions Setup
🧬 Try It Out!
Debugging & Error Handling
Let's examine our code with line numbers to understand debugging
Understanding Python Error Messages
🔍 Anatomy of a Python Error
🐛 Example 1: Syntax Error
Missing parentheses - Python can't understand the code structure
🧮 Example 2: Type Error
Trying to use incompatible data types together
🏷️ Example 3: Name Error
Using a variable or function that doesn't exist
💡 Debugging Tips
- • Start from the bottom - that's the actual error
- • Note the line number and file name
- • Look at the exact code line mentioned
- • SyntaxError: Check parentheses, quotes, colons
- • TypeError: Check if data types match
- • NameError: Check spelling and definitions
Defensive Programming for Biological Data
🛡️ Expect the Unexpected
Real biological data is messy:
- • DNA sequences might not contain ATG start codons
- • FASTA files may have ambiguous bases (N, R, Y)
- • User input could be empty or invalid
- • Sequences might be too short for analysis
Solution: Use if statements to validate data and handle expected scenarios gracefully!
🔍 Simple ATG Finder with Data Validation
Two key checks: valid DNA bases and ATG presence
💡 Defensive Programming Principles
- • Check for empty or None values
- • Validate data types (string vs number)
- • Verify biological constraints
- • No ATG found → return None
- • Invalid bases → clean or warn
- • Short sequences → inform user
- • Print warnings for data issues
- • Return meaningful values
- • Document what went wrong
- • Return None instead of crashing
- • Continue processing when possible
- • Don't let one bad sequence stop analysis
🔄 Alternative: try/except blocks
try/except is useful for building robust software applications:
- • File I/O operations
- • Network connections
- • Database queries
- • User interface errors
- • Use simple
ifstatements - • Validate data explicitly
- • Handle expected scenarios
- • Focus on data quality
💡 For data science: Missing ATGs, invalid bases, or empty sequences aren't "exceptions" - they're normal biological scenarios that need explicit handling with if statements.
DNA Sequence File Formats: FASTA
📄 What is a FASTA File?
FASTA is the most common format for storing DNA, RNA, and protein sequences. It's a simple text format that biologists use worldwide.
💡 Fun Fact
FASTA was named after the FASTA software program for sequence alignment, developed in the 1980s at the University of Virginia
🏗️ FASTA Format Structure
Basic Format Rules
- • Header line starts with
> - • Sequence ID comes right after
> - • Description (optional) after the ID
- • Sequence data on following lines
- • No line length limit for sequence
Example FASTA File
>NM_000546.6 Homo sapiens tumor protein p53ATGGAGGAGCCGCAGTCAGATCCTAGCGTCGAGCCCCCTCTGAGTCAGGAAACATTTTCAGACCTATGGAAACTACTTCCTGAAAACAACGTTCTGTCCCCCTTGCCGTCCCAAGCAATGGATGATTTGATGCTGTCCCCGGACGATATTGAACAATGGTTCACTGAAGACCCAGGTCCAGATGAAGCTCCCAGAATGCCAGAGGCTGCTCCCCG
>NM_001126115.2 Homo sapiens BRCA1 geneATGGATTTCCGTCTGAACAAACAACACCGCCGGCCCCGTGGGTCCGTGTCCCCGGCAAGCCCCACCCGGGCCCTCCCTCCCGGCTGGGGGCCGCCCCCCGACACCAATCAGGCCCCCCACCCCGGCTCTCTACCCCCGCGCCCCCGGACACTACCCCCCGCC🌐 Where to Find FASTA Files
NCBI GenBank
National Center for Biotechnology Information
- • Comprehensive gene database
- • Download individual genes
- • Genome assemblies available
Ensembl
European genome annotation database
- • High-quality annotations
- • Multiple species genomes
- • Easy bulk downloads
UniProt
Protein sequence database
- • Protein sequences only
- • Functional annotations
- • Research-quality curation
💡 Key Takeaways: Working with FASTA Files
FASTA Essentials
- • Simple, universal sequence format
- • Header starts with
> - • Can contain multiple sequences
- • Used by all major databases
Python Skills
- • File reading with
open() - • String manipulation for parsing
- • Dictionary storage for multiple sequences
- • Always handle the last sequence!
🎯 FASTA files are your gateway to analyzing real biological sequences!
💻 FASTA File Parsing: Professional Approach
Here's how bioinformaticians parse FASTA files in the real world - handling multiple sequences and complex structures.
🧬 Complete FASTA Parser
sequences = {}current_gene_id = Nonecurrent_sequence = ""
# Use context manager to safely open and read the filewith open(filename, 'r') as file: for line in file: line = line.strip() # Remove whitespace
if line.startswith('>'): # Save previous sequence if we have one if current_gene_id is not None: sequences[current_gene_id]['sequence'] = current_sequence
# Parse new header header_parts = line[1:].split(' ', 1) # Split on first space only current_gene_id = header_parts[0]
# Store header info sequences[current_gene_id] = { 'header': line[1:], # Full header without > 'sequence': "" }
current_sequence = "" # Reset sequence print(f"Found sequence: {current_gene_id}")
else: # Add to current sequence (sequences can span multiple lines) current_sequence += line.upper()
# Don't forget the last sequence!if current_gene_id is not None: sequences[current_gene_id]['sequence'] = current_sequence
# Display resultsfor gene_id, data in sequences.items(): print(f"\nGene: {gene_id}") print(f"Header: {data['header']}") print(f"Length: {len(data['sequence'])} bases") print(f"First 50 bases: {data['sequence'][:50]}...")🔍 Why This Approach Works
🧩 Handles Multiple Sequences
Real FASTA files often contain multiple sequences. This parser stores each one with its own ID and metadata.
📏 Line-by-Line Processing
Sequences can span multiple lines. This approach reads line by line and concatenates sequence data properly.
💾 Smart Data Structure
Uses nested dictionaries to store both header information and sequence data for easy access.
⚠️ Edge Case Handling
Don't forget the last sequence! The final sequence needs special handling since there's no next header.
💡 Key Programming Concepts
- • Context Managers -
with open()safely handles files - • String Methods -
.strip(),.startswith(),.split() - • State Management - Tracking current sequence and ID
- • Nested Dictionaries - Complex data organization
- • Edge Cases - Handling the last sequence properly
- • Data Validation - Checking for None values
🎓 Ready for More Advanced Practice?
Build complete FASTA parsers and work with real research datasets
Advanced FASTA AnalysisFile I/O & the with Statement
📁 Why File Handling Matters in Biology
Biological data lives in files - sequences, experiment results, annotations. Learning proper file handling is essential for any bioinformatics work.
🧬 Real Examples
FASTA sequences, CSV experiment data, JSON annotations, XML databases, TSV gene expression data, and many more!
⚠️ The Problem: Files Can Get "Stuck Open"
❌ The Old Way (Risky)
# Opening a file the old wayfile = open('sequences.fasta', 'r')content = file.read()# Process the content...
# What if an error happens here?# The file might never get closed!# This can cause problems...
file.close() # Might never execute!🚨 What Can Go Wrong
- • Memory leaks - Files stay open in memory
- • File locks - Other programs can't access the file
- • Resource exhaustion - System runs out of file handles
- • Data corruption - Writes might not be saved
- • Crashes - Program errors leave files open
✅ The Solution: Context Managers & with Statement
💚 The Safe Way
# Using the with statementwith open('sequences.fasta', 'r') as file: content = file.read() # Process the content...
# Even if an error happens here, # the file will ALWAYS be closed!
# File is automatically closed here# No matter what happened above!🎯 Why It's Better
- • Automatic cleanup - Files always close
- • Exception safe - Works even if errors occur
- • Cleaner code - No need to remember .close()
- • Best practice - Used by all professional developers
- • Resource efficient - Prevents memory leaks
💡 Key Takeaways: File I/O Best Practices
Essential Rules
- • Always use
with open() - • Choose the right file mode for your task
- • Handle large files line by line
- • Check if files exist before reading
Bioinformatics Tips
- • Use
.strip()to remove whitespace - • Process files line by line for memory efficiency
- • Validate file formats before processing
- • Always backup important data files
🎯 Proper file handling prevents data loss and makes your code more reliable!
Lecture 2 Summary: What You've Learned Today
1️⃣String Operations & DNA Analysis
String Slicing
- ▸Extract sequence parts:
dna[0:3] - ▸Find reading frames and ORFs
String Methods
- ▸
.find(),.upper(),.replace() - ▸Search for start/stop codons
Biological Context
- ▸Reading frames and translation
- ▸Open Reading Frame analysis
2️⃣Conditionals & Decision Making
If Statements
- ▸
if condition: - ▸Make decisions in code
Logical Operators
- ▸
and,or,not - ▸Complex condition testing
Error Handling
- ▸Validate input sequences
- ▸Defensive programming
3️⃣Dictionaries & Data Organization
Key-Value Pairs
- ▸
{'codon': 'amino_acid'} - ▸Store genetic code tables
Translation
- ▸DNA → RNA → Protein
- ▸Codon table lookups
File Handling
- ▸FASTA file parsing
- ▸
with open()best practices
🧬Real Bioinformatics Applications
ORF Finding
Identify potential protein-coding regions in DNA sequences
Sequence Translation
Convert DNA to protein sequences using codon tables
File Processing
Parse and analyze biological data formats like FASTA
💡Advanced Programming Skills Gained
- ✓String manipulation for sequence analysis
- ✓Data validation with conditionals
- ✓Efficient data lookup using dictionaries
- ✓File I/O operations for real data
- ✓Error handling and defensive coding
- ✓Biological data processing workflows
🚀What's Coming Next
Lecture 3
Data Analysis with Pandas: Tables, Gene Dependencies & Correlation
Pandas DataFrames
Work with tabular biological data, CSV files, and gene expression datasets
Gene Dependencies
Analyze relationships between genes, correlations, and biological networks
Data Visualization
Create plots and charts to visualize biological data patterns
🎉 Excellent Progress!
You can now analyze DNA sequences, parse biological files, and make data-driven decisions in code
Next week we'll explore how to work with large datasets and discover gene relationships using Python!
Resources for DNA Pythonistas 🐍🧬
Biopython
The essential Python library for biological computation
What Biopython Does
- ▸Parse biological file formats (FASTA, GenBank, PDB, etc.)
- ▸Sequence manipulation and translation
- ▸BLAST searches and alignment tools
- ▸Access NCBI databases (Entrez, PubMed)
- ▸Phylogenetic tree analysis
Getting Started
Install with pip:
pip install biopythonQuick example:
from Bio.Seq import Seq
dna = Seq("ATGGCCATTGTAA")
protein = dna.translate()
print(protein) # MAIV*🧰Other Essential Python Libraries for Biology
NumPy & Pandas
Numerical computing and data analysis. Essential for working with gene expression data, experimental results, and large datasets.
numpy.org • pandas.pydata.orgscikit-bio
Bioinformatics library for sequence alignment, diversity analysis, and working with biological data structures.
scikit-bio.orgmatplotlib & seaborn
Data visualization libraries for creating publication-quality plots, charts, and figures for your biological data.
matplotlib.org📖Learning Resources & Documentation
Online Courses & Tutorials
- ▸Python for Biologists
pythonforbiologists.com - Comprehensive tutorials
- ▸Rosalind
rosalind.info - Learn bioinformatics through problem solving
- ▸BioPython Tutorial
Official tutorial with real-world examples
Databases & APIs
- ▸NCBI Entrez
Access GenBank, PubMed, and other NCBI databases via Python
- ▸UniProt
Protein sequence and functional information database
- ▸Ensembl REST API
Genomic data access through Python requests
🔬Specialized Bioinformatics Tools
PyMOL
3D molecular visualization and analysis of protein structures
DendroPy
Phylogenetic computing library for tree analysis
pysam
Python wrapper for SAM/BAM sequencing data formats
💬Community & Getting Help
Bioinformatics StackExchange
Ask questions, get answers from the community
GitHub
Explore open-source bioinformatics projects
Python Documentation
Official Python docs - your best friend!
🚀 You're Now Part of the DNA Pythonista Community!
These tools will empower you to tackle real biological problems with code
Start exploring Biopython today and see how much time you can save in your research!